Colorectal Cancer Research at the Goodman Cancer Institute: A long-standing quest
March 17 2025

Rosalind and Morris Goodman Cancer Institute (GCI) emerita professor Nicole Beauchemin, PhD, has played an integral role in groundbreaking colorectal cancer research conducted at the GCI. Read her article below to learn more about the GCI's legacy of advancing colorectral cancer research.
Colorectal cancer (CRC) has been a long-standing research quest at the Goodman Cancer Institute ever since Dr. Phil Gold, the founder of the original McGill Cancer Centre, began his research projects to identify tumor-specific antigens serving as biomarkers, to follow the evolution of the disease or as targets for specific therapies. This led to the identification of Carcinoembryonic antigen (CEA) and the further use of specific monoclonal antibodies as detection tools in clinical settings (1). CEA overexpression in tumors versus normal tissues with specific anti-CEA antibodies demonstrated very high sensitivity (2). Preoperative serum CEA levels were elevated when tested in a cohort of >17,000 patients; this test confirmed a 60% increased risk of overall mortality at all stages of the cancer (3). Thus, CEA has become an excellent biomarker used clinically worldwide in monitoring CRC progression. In addition, characterization of the CEA cDNA in 1987 by several international teams, including the Stanners group produced an unexpected finding in that CEA was the hallmark of a large gene family (the CEACAM gene family) with as many as 22 family members, all bearing characteristic Immunoglobulin-like domains (4).
The three most important CEACAM family members involved in colorectal cancer are CEA, CEACAM6 and CEACAM1 (Figure 1). All of them function as intercellular adhesion molecules (4). In addition to CEA, further studies confirmed CEACAM6 also plays a role in CRC progression, its expression being up-regulated from adenomas to malignant tumors; its enhanced expression is statistically associated with poorer overall survival and disease-free survival (5). Chan and Stanners (GCI) developed a CEABAC transgenic mouse model, harboring a bacterial artificial chromosome containing the human CEA, CEACAM6 and CEACAM7 genes that overexpressed CEA and CEACAM6 proteins from 2-20 fold in the colon. This led to striking colorectal tissue disorganization (hyperplasia and dysplasia) and when CRC cancer was provoked with the Azoxymethane carcinogen, colon tumors were more prominent in numbers and size as compared to those appearing in control mice (Figure 1) (6).
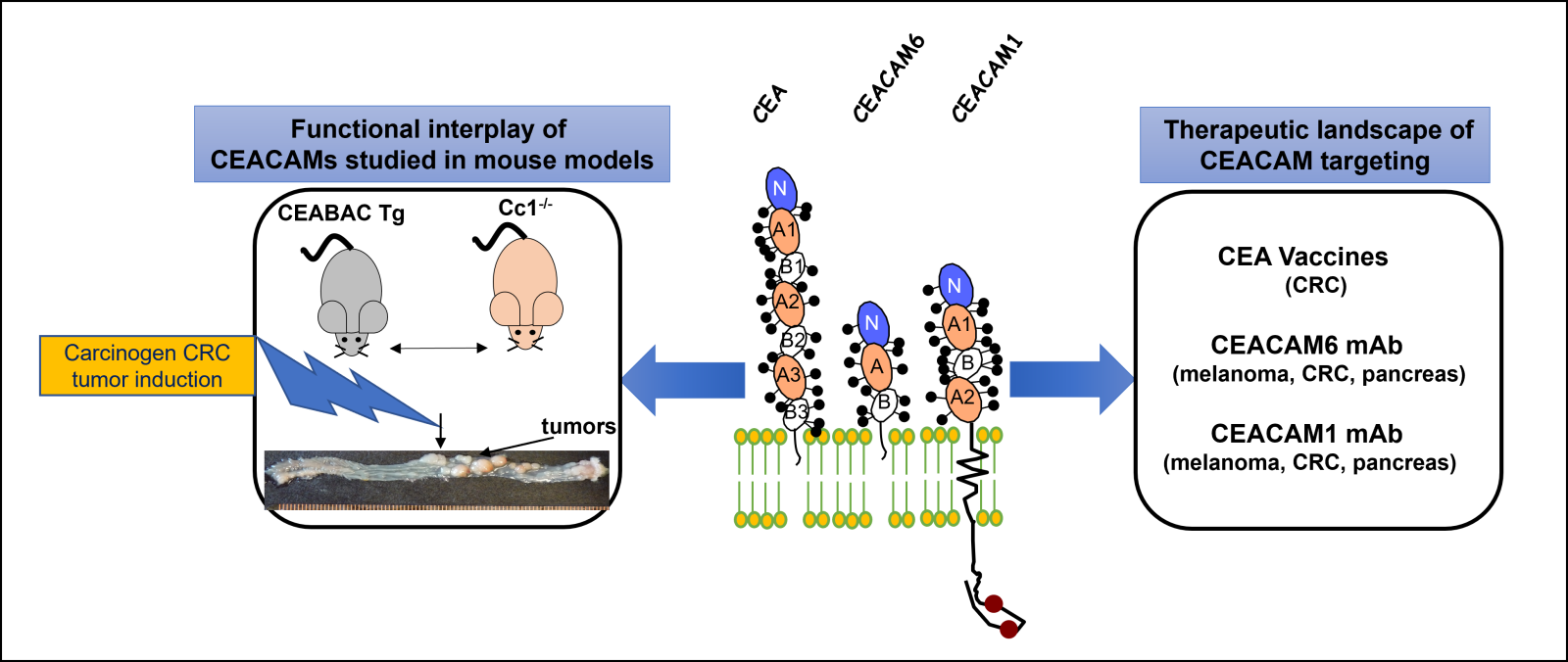 Figure 1: Colorectal cancer (CRC) has been widely studied at the Goodman Cancer Institute over the years. These studies have led to the identification of the Carcinoembryonic Antigen (CEA), a widely used CRC biomarker, but also to a large gene family of the CEACAM proteins (CEA Cell Adhesion Molecules) of which CEACAM6 and CEACAM1 are also key players in the development/progression of CRC (CENTRAL PANEL). The CEACAM proteins exhibit Immunoglobulin domains and are highly glycosylated (sticks) with either a phosphatidyl-inositol linkage (CEA and CEACAM6) or a transmembrane and a long cytoplasmic domain (CEACAM1) with two Tyr-phosphorylatable residues (red balls). To understand the roles of CEA, CEACAM6 and CEACAM1 in CRC progression, several mouse models have been developed either overexpressing the CEA, CEACAM6 and CEACAM7 genes (the CEABAC mouse) or the CEACAM1-deficient (Cci1-/- mouse). When treated with a CRC-specific carcinogen inducer, CRC tumors develop and are assessed (LEFT PANEL). Over the years, many therapeutic approaches have been studied by the international CEACAM research groups that are presently still undergoing clinical validation (RIGHT PANEL).
Figure 1: Colorectal cancer (CRC) has been widely studied at the Goodman Cancer Institute over the years. These studies have led to the identification of the Carcinoembryonic Antigen (CEA), a widely used CRC biomarker, but also to a large gene family of the CEACAM proteins (CEA Cell Adhesion Molecules) of which CEACAM6 and CEACAM1 are also key players in the development/progression of CRC (CENTRAL PANEL). The CEACAM proteins exhibit Immunoglobulin domains and are highly glycosylated (sticks) with either a phosphatidyl-inositol linkage (CEA and CEACAM6) or a transmembrane and a long cytoplasmic domain (CEACAM1) with two Tyr-phosphorylatable residues (red balls). To understand the roles of CEA, CEACAM6 and CEACAM1 in CRC progression, several mouse models have been developed either overexpressing the CEA, CEACAM6 and CEACAM7 genes (the CEABAC mouse) or the CEACAM1-deficient (Cci1-/- mouse). When treated with a CRC-specific carcinogen inducer, CRC tumors develop and are assessed (LEFT PANEL). Over the years, many therapeutic approaches have been studied by the international CEACAM research groups that are presently still undergoing clinical validation (RIGHT PANEL).
Our own group focused its research on the CEACAM1 protein, another member of the same family but with very different properties and broader expression patterns: it is present on many tumor types including CRC, but also on the surface of small blood vessels, lymph nodes and many immune cell types (4). CEACAM1 is also a cell adhesion protein, but it projects into the cytoplasm one of two cytoplasmic domains with one longer one (CEACAM1-L) shown to be Tyrosine-phosphorylated by a variety of Tyr kinases; this leads to high affinity interactions with Tyr phosphatases SHP-1 and SHP-2 and activation of many signaling cascades (4). In human CRC development, CEACAM1 expression was initially reported as downregulated (contrary to upregulation of CEA and CEACAM6) in adenomas, the early phase of colorectal cancer. To confirm that its downregulation was significant in CRC development, we developed a Ceacam1-/- mouse model; CEACAM1 deletion led to increased colon proliferation compared with controls, with the colonic cells showing decreased programmed cell death. Upon CRC cancer triggering through azoxymethane carcinogen treatment, the Ceacam1-null mice produced a great number of colon tumors (7). In a second CRC model, the Ceacam1-/- X Apc1638N/+ mouse model, elimination of CEACAM1 expression increased CRC tumors in size and numbers (8). However, recent clinical data indicate that CEACAM1 increases with the level of colon dysplasia (9); it remains possible that the balance between short and long cytoplasmic domains of CEACAM1 plays a significant role in this discrepancy (9). Paradoxically, advanced stages of CRC exhibit high CEACAM1 expression which is associated with invasiveness and liver metastasis (10-13).
With the advent of immunotherapy as an effective anti-cancer therapy, CEACAM1 was recognized as an anti-cancer clinical target in line with its prominent immune cell expression. It is present at the surface of circulating CD8+ T lymphocytes and CRC Tumor-Infiltrating Lymphocytes as well as other immune cells (14). Availability of a highly specific monoclonal antibody recognizing only the CEACAM1 member (exclusive of all other CEACAMs) has shown that blocking CEACAM1 activity with this antibody stops the development of and reverses CRC and other cancers; inhibition of CEACAM1 activity disengages it from physical contact with two other important regulators of immune cells, namely TIM-3 and PD-1 (15). CEACAM1 inhibition releases the brake on T cells imposed by TIM-3 or PD-1 and reactivates these T cells to combat many different forms of cancers, including CRC (14, 15). Future clinical trials may validate its usefulness to combat cancer progression in human patients. Clinical translation in this area has been slowed by the lack of knowledge of how these three CEACAM family members interact with each other on the cell surface of normal and malignant CRC cells, but also between those expressed on tumor cells and tumor-infiltrating immune cells (16, 17).
Philippe Gros’ group has also made significant strides in understanding another form of the disease, colitis-associated CRC (CA-CRC) by identifying susceptibility gene loci in mouse models (Ccs3 and Ccs5) and the interferon regulatory factor 1 gene (Irf1) (18,19). Deletion of Irf1 provokes significant inflammation and infiltration of myeloid, granulocytic and lymphoid immune cell into mouse colons with significant tumor development when triggered by carcinogen treatments. The same transcriptional changes associated with mouse Irf1 deletion are found in human clinical specimens of stage 3 and 4 CRC cancers and associated with a poorer prognosis (19). These results highlight many of the possible biomarkers used to assess the progression of CRC in patients, to offer timely and appropriate aggressive patient treatments.
Article authored by Nicole Beauchemin, PhD.
References: (bolded names correspond to members of the Rosalind and Morris Goodman Cancer Institute)
1. Gold P, Freedman SO. (1965); PMID: 4953873
2. Tiernan, J.P. et al., (2013); PMID; 23322207
3. Thirunavukarasu, P. et al. (2011); PMID: 21421861
4. Beauchemin, N., and Arabzadeh, A. (2013); PMID: 23903773
5. Guanhua Wu, et al. (2024); PMID: 38240103
6. Chan CH, Cook D, Stanners CP. (2006); PMID: 16632476
7. Leung, N….Beauchemin N. (2006); PMID: 16619040
8. Leung, N….Beauchemin N. (2008); PMID: 18454175
9. Götz, L et al. (2024); PMID: 39674877
10. Ieda, J. et al. (2011); PMID: 21413011
11. Yeatman, T.J. et al. (1997); PMID:9834361
12. Kang, W.Y. et al. (2007); PMID:17143599
13. Song, J. H. et al. (2011); PMID:20524097
14. Huang, Y.H……Beauchemin, N., Blumberg, R.S. (2024); PMID: 38956268
15. Huang, Y.H ….Beauchemin, N. …Blumberg, R.S. (2015); PMID: 25363763
16. Stern, N. et al. (2005); PMID: 15905509
17. Pinkert, J. et al. (2022); PMID: 35141051
18. Meunier, C……Beauchemin, N., Gros, P. (2010); PMID: 19915610
19. Jeyakumar, T. ….Beauchemin, N., Gros, P. (2019); PMID: 31827213
Rosalind & Morris Goodman Cancer Institute all rights reserved • Privacy policy • Accessibility
Website by Riposte